Research
Try real-time localization of acupuncture points AI (https://attention.pknu.ac.kr:18501)
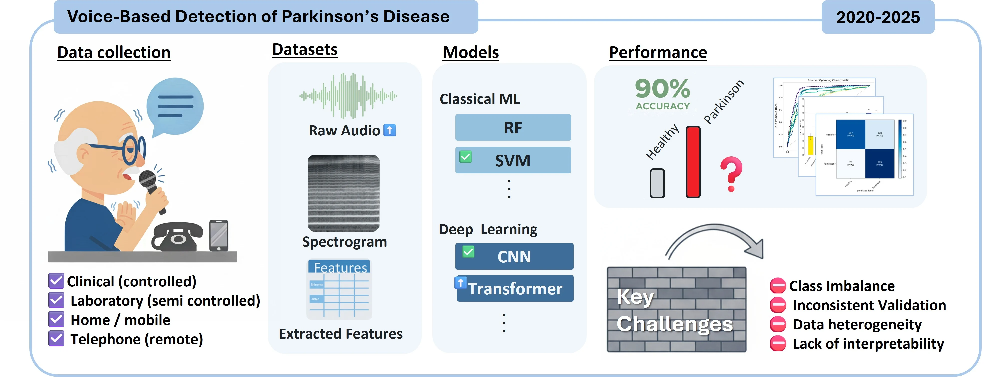

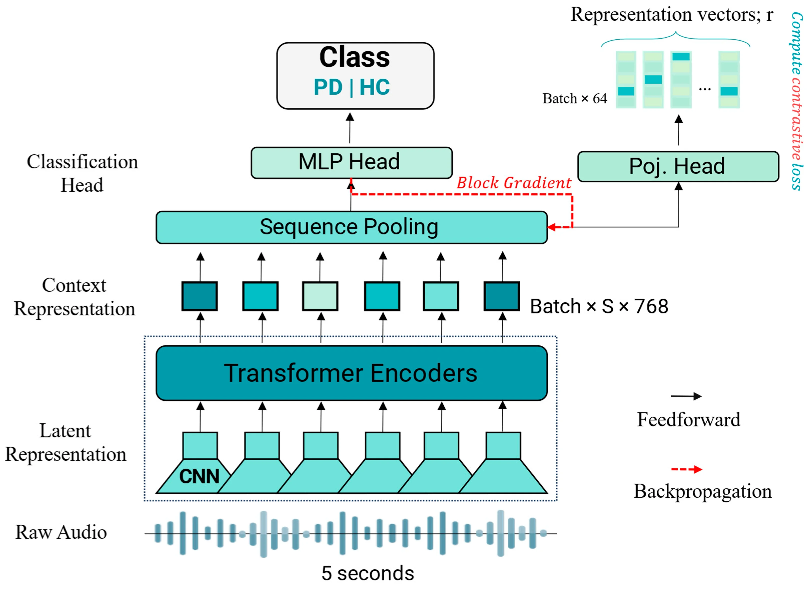
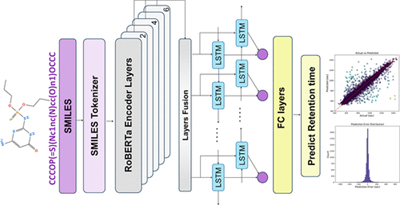
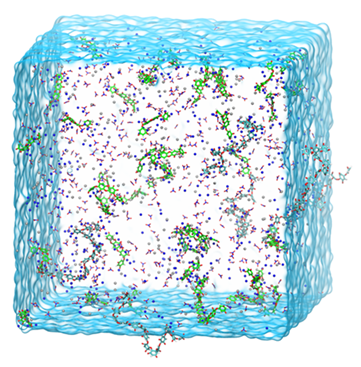




Real-time Location of Acupunture Points Based on Anatomical Landmarks and Pose Estimation Models




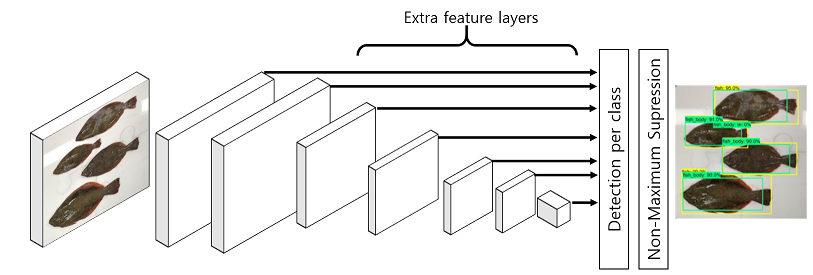
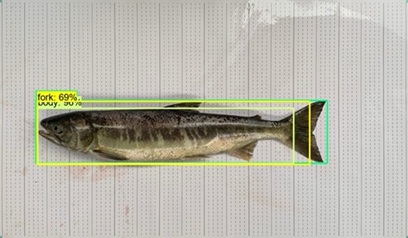
Automated meaturement of fish lengths
Gating Mechanism of the Voltage-Gated Proton Channel Studied by Molecular Dynamics Simulations

Functional stability of water wire-carbonyl interactions in an ion channel



Silver-Lactoferrin Nanocomplexes as a Potent Antimicrobial Agent




Insight into the mechanism of the influenza A proton channel from a structure in a lipid bilayer (PDB deposit: 2L0J).


Conformational heterogeneity of the M2 proton channel and a structural model for channel activation.

.png")



